Home » OA/TOF information » OA/TOF Information for Healthcare Professionals » Adult OA/TOF Management Handbook
Home » OA/TOF information » OA/TOF Information for Healthcare Professionals » Adult OA/TOF Management Handbook
Written by Dr Caroline Love / current version 1.1 (Sep, 2022)
Attention healthcare professionals
We are happy to offer healthcare professionals a free digital copy of both “The TOF Book” and “The Soft Food Recipe Book” along with all our resources.
Share with your GP

Many adults born with OA/TOF still have ongoing sequelae from their childhood anomalies. In the early days of repair, parents were told they were ‘fixed’ and should live an entirely normal life. However, as these babies have grown up into middle-aged adults, research consistently demonstrates that a repaired oesophagus and trachea do not function like an oesophagus and trachea that developed in normal continuity in utero. Oesophageal nerve and muscle do not function normally in almost all adults born with OA/TOF, and dysphagia, gastro-oesophageal reflux disease (GORD) and laryngopharyngeal reflux (LPR) are common as a result of this, though not universal, and many with these issues may not realise as they have never had a normal oesophagus. Similarly, alongside the TOF formation, the C shaped cartilage may not form properly, leaving tracheomalacia (TM) as a result, which can lead to ongoing respiratory problems.
However, whilst there is good awareness of health problems related to OA/TOF amongst paediatric surgeons who treat the condition, when these children transition to adult services, adults struggle to find doctors in primary and secondary care who are knowledgeable about the anomaly. Some adults are reluctant to seek medical opinions on their health issues due to negative experiences and lack of awareness in the past. Others have become their own expert patient but need the support of an advocate GP.
With this handbook, we hope that we can fill in the knowledge gap of a rare and complex condition to help GPs understand, treat and signpost treatment to the appropriate specialist when needed. It is impossible for GPs to be fully informed about the near-infinite number of rare diseases that exist, but since there are only a handful of tertiary care adult surgeons and physicians across the UK with experience in management of adults born with OA/TOF, this handbook aims to be a management handbook for primary care developed from the most recent research in this area.
Many adults born with OA/TOF seeking GP appointments have issues related to the condition that are commonly treatable in primary care, such as GORD, LPR, sinusitis; however the TOFS charity does have a list of experienced tertiary care doctors for when this is appropriate, and these doctors are usually happy to be written to/emailed with queries by GPs as well as referrals when needed.
Oesophageal atresia (OA) and tracheo-oesophageal fistula (TOF) are two congenital anomalies that occur in around 1/2,600 births. (1) Whilst the two can arise separately, they commonly arise together as the trachea and oesophagus develop from the separation of a common foregut early in foetal development. If this foregut fails to completely develop, and/or separate, then the anomalies arise. (2)
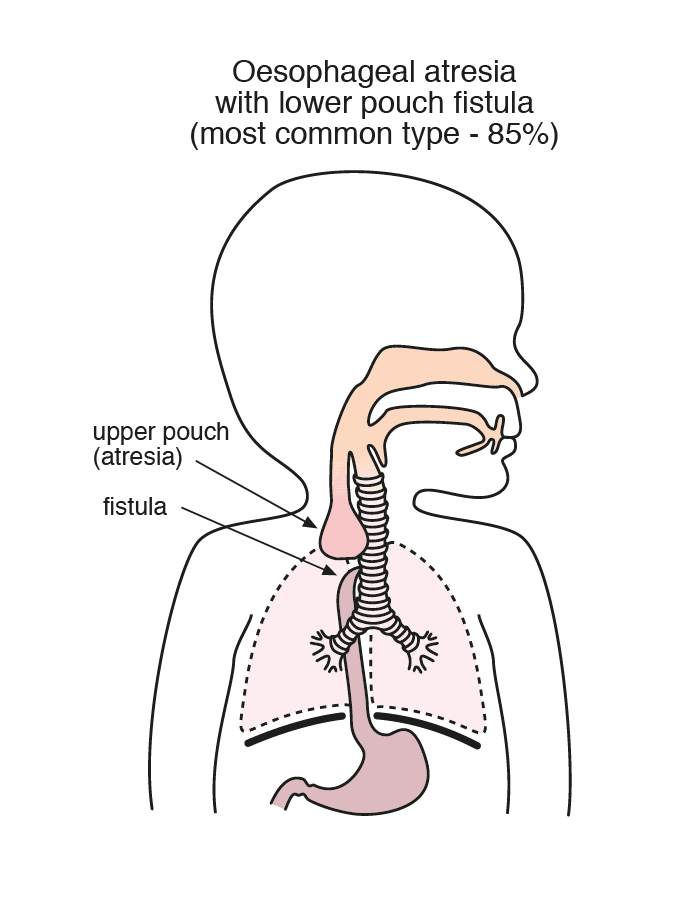
The first successful surgical repair of the anomaly was performed in 1941, and survival is now consistently over 90% worldwide post repair. Survival is almost universal (98.9%) in infants born today with the condition at a birthweight over 1.5kg and no cardiac anomaly, 82% for those weighing either <1.5kg or with a cardiac anomaly, but only 50% for those infants with both low birth weight and cardiac anomaly, based on Great Ormond Street Hospital data. (3) Gross devised the most commonly used classification system in 1953, dividing the anomaly into five main types.
A further phenotypic separation, and the one that has the greatest ramifications for ongoing morbidity in childhood and adulthood, is the length of separation of the gap between distal and proximal oesophagus.
Atresia gap less than two vertebrae at birth. These can have primary repair (PR) of the oesophagus (anastomosis of the two ends together shortly after birth). Currently this is almost universally performed by thoracotomy or increasingly endoscopically, but in the past many current adults may have had other surgical approaches, including substernal.
When the gap is larger than two vertebrae, the closure may be delayed to allow baby and oesophagus to grow, at which point delayed primary closure is performed or, if it is larger still ‘long gap’ (six vertebrae plus), oesophageal replacement may be needed, such as gastric pull-up/transposition (as the name implies, the stomach is ‘pulled up’ to connect with the proximal end of the oesophagus), jejunal or colonic transposition (where a section of the jejunum or colon is used to bridge the gap between the two ends of the oesophagus).
Long-gap OA patients spend much longer in Neonatal Intensive Care Unit (NICU)under anaesthetic, in hospital before discharge and may need several operative procedures before repair is complete. They also are more likely to need multiple admissions in childhood, need nutritional support and have ongoing OA/TOF morbidity in childhood and adulthood.
Many adults born with long-gap OA spent their first year of life in hospital, with all the impact that thus follows on development, family life and attachment, and medical trauma. However, as the rest of the leaflet will demonstrate, short-gap OA/TOF also leaves a lifelong impact on the body.
Over 50% of those born with OA/TOF have additional anomalies, cardiovascular in 29%, anorectal in 14%, genitourinary in 14%, additional gastrointestinal in 13%, skeletal in 10%, respiratory in 6%, genetic in 4% and other anomalies outside the above in 11%. This is most frequently the case in those with pure OA, and least common in Type C OA/TOF. OA/TOF forms the T and E of the VACTERL association of anomalies, and is also associated with CHARGE syndrome. (3) As is the case with OA/TOF, sometimes these additional anomalies become more serious in adult life.
| Cookie | Duration | Description |
|---|---|---|
| cookielawinfo-checkbox-advertisement | 1 year | Set by the GDPR Cookie Consent plugin, this cookie is used to record the user consent for the cookies in the "Advertisement" category . |
| cookielawinfo-checkbox-analytics | 1 year | Set by the GDPR Cookie Consent plugin, this cookie is used to record the user consent for the cookies in the "Analytics" category . |
| cookielawinfo-checkbox-functional | 1 year | The cookie is set by the GDPR Cookie Consent plugin to record the user consent for the cookies in the category "Functional". |
| cookielawinfo-checkbox-necessary | 1 year | Set by the GDPR Cookie Consent plugin, this cookie is used to record the user consent for the cookies in the "Necessary" category . |
| cookielawinfo-checkbox-others | 1 year | Set by the GDPR Cookie Consent plugin, this cookie is used to store the user consent for cookies in the category "Others". |
| cookielawinfo-checkbox-performance | 1 year | Set by the GDPR Cookie Consent plugin, this cookie is used to store the user consent for cookies in the category "Performance". |
| CookieLawInfoConsent | 1 year | Records the default button state of the corresponding category & the status of CCPA. It works only in coordination with the primary cookie. |
| elementor | never | This cookie is used by the website's WordPress theme. It allows the website owner to implement or change the website's content in real-time. |
| enforce_policy | 1 year | PayPal sets this cookie for secure transactions. |
| ts | 3 years | PayPal sets this cookie to enable secure transactions through PayPal. |
| ts_c | 3 years | PayPal sets this cookie to make safe payments through PayPal. |
| Cookie | Duration | Description |
|---|---|---|
| aka_debug | session | Vimeo sets this cookie which is essential for the website to play video functionality. |
| nsid | session | This cookie is set by the provider PayPal to enable the PayPal payment service in the website. |
| player | 1 year | Vimeo uses this cookie to save the user's preferences when playing embedded videos from Vimeo. |
| tsrce | 3 days | PayPal sets this cookie to enable the PayPal payment service in the website. |
| x-pp-s | session | PayPal sets this cookie to process payments on the site. |
| Cookie | Duration | Description |
|---|---|---|
| l7_az | 30 minutes | This cookie is necessary for the PayPal login-function on the website. |
| sync_active | never | This cookie is set by Vimeo and contains data on the visitor's video-content preferences, so that the website remembers parameters such as preferred volume or video quality. |
| Cookie | Duration | Description |
|---|---|---|
| _ga | 2 years | The _ga cookie, installed by Google Analytics, calculates visitor, session and campaign data and also keeps track of site usage for the site's analytics report. The cookie stores information anonymously and assigns a randomly generated number to recognize unique visitors. |
| _gat_UA-51564864-7 | 1 minute | A variation of the _gat cookie set by Google Analytics and Google Tag Manager to allow website owners to track visitor behaviour and measure site performance. The pattern element in the name contains the unique identity number of the account or website it relates to. |
| _gcl_au | 3 months | Provided by Google Tag Manager to experiment advertisement efficiency of websites using their services. |
| _gid | 1 day | Installed by Google Analytics, _gid cookie stores information on how visitors use a website, while also creating an analytics report of the website's performance. Some of the data that are collected include the number of visitors, their source, and the pages they visit anonymously. |
| _hjAbsoluteSessionInProgress | 30 minutes | Hotjar sets this cookie to detect the first pageview session of a user. This is a True/False flag set by the cookie. |
| _hjFirstSeen | 30 minutes | Hotjar sets this cookie to identify a new user’s first session. It stores a true/false value, indicating whether it was the first time Hotjar saw this user. |
| _hjIncludedInPageviewSample | 2 minutes | Hotjar sets this cookie to know whether a user is included in the data sampling defined by the site's pageview limit. |
| _hjIncludedInSessionSample | 2 minutes | Hotjar sets this cookie to know whether a user is included in the data sampling defined by the site's daily session limit. |
| CONSENT | 2 years | YouTube sets this cookie via embedded youtube-videos and registers anonymous statistical data. |
| vuid | 2 years | Vimeo installs this cookie to collect tracking information by setting a unique ID to embed videos to the website. |
| Cookie | Duration | Description |
|---|---|---|
| VISITOR_INFO1_LIVE | 5 months 27 days | A cookie set by YouTube to measure bandwidth that determines whether the user gets the new or old player interface. |
| YSC | session | YSC cookie is set by Youtube and is used to track the views of embedded videos on Youtube pages. |
| yt-remote-connected-devices | never | YouTube sets this cookie to store the video preferences of the user using embedded YouTube video. |
| yt-remote-device-id | never | YouTube sets this cookie to store the video preferences of the user using embedded YouTube video. |
| yt.innertube::nextId | never | This cookie, set by YouTube, registers a unique ID to store data on what videos from YouTube the user has seen. |
| yt.innertube::requests | never | This cookie, set by YouTube, registers a unique ID to store data on what videos from YouTube the user has seen. |
| Cookie | Duration | Description |
|---|---|---|
| _hjSession_2528865 | 30 minutes | No description |
| _hjSessionUser_2528865 | 1 year | No description |
| LANG | 9 hours | No description |